A facile technology for the high-throughput sequencing of the paired VH:VL and TCRβ:TCRα repertoires – Science Advances
Abstract
Natively paired sequencing (NPS) of B cell receptors [variable heavy (VH) and light (VL)] and T cell receptors (TCRb and TCRa) is essential for the understanding of adaptive immunity in health and disease. Despite many recent technical advances, determining the VH:VL or TCRb:a repertoire with high accuracy and throughput remains challenging. We discovered that the recently engineered xenopolymerase, RTX, is exceptionally resistant to cell lysate inhibition in single-cell emulsion droplets. We capitalized on the characteristics of this enzyme to develop a simple, rapid, and inexpensive in-droplet overlap extension reverse transcription polymerase chain reaction method for NPS not requiring microfluidics or other specialized equipment. Using this technique, we obtained high yields (5000 to >20,000 per sample) of paired VH:VL or TCRb:a clonotypes at low cost. As a demonstration, we performed NPS on peripheral blood plasmablasts and T follicular helper cells following seasonal influenza vaccination and discovered high-affinity influenza-specific antibodies and TCRb:a.
INTRODUCTION
Immune receptors [B and T cell receptors (BCRs and TCRs)] play essential roles in adaptive immunity, and thus enumerating and identifying these sequences are key to understanding responses to infection or vaccination, for discovering therapeutic antibodies, and for engineering T cells (1–3). Antibodies are composed of variable heavy (VH) and light (VL) chains, and TCRs are composed of TCRα/γ and TCRβ/δ chains. Antibodies are subsequently subjected to somatic hypermutation (SHM) in germinal centers, a process required for affinity maturation and antigen specificity (2). Because the two chains of adaptive immune receptors are encoded by two transcripts, determining the BCR or TCR repertoire necessitates sequencing of two amplicons from each cell. Early natively paired sequencing (NPS) approaches relied on limiting dilution of lymphocytes, followed by reverse transcription polymerase chain reaction (RT-PCR) and Sanger sequencing; although widely adopted, these techniques are inherently limited to the analysis of at most a few hundred cells (2, 4, 5), because they are costly and not readily scalable. Combinatorial pairing (pairSEQ) has been used for NPS of TCRs; however, it is not suitable for detecting both rare clones and abundant clones in the same sample, and furthermore, it has not been used for antibody repertoire studies (6). Single-cell, droplet-based RT-PCR techniques have the potential to meet the very high throughput (>105 B or T lymphocytes) desired for biomedical studies (7). However, because the high concentration of cell lysate components within the miniscule volume of typical emulsion droplets markedly inhibits the RT-PCR (8–10), one-step cell emulsification techniques, while straightforward, are very inefficient, are plagued by very low yields of paired receptors, and result in a highly skewed repertoire in which a single clonotype accounts for >25% of the reads (11). To circumvent this limitation, continuous flow systems and microfluidic devices have been used to encapsulate cells in larger droplets [thus, resulting lower cell lysate concentrations and more efficient complementary DNA (cDNA) synthesis] coupled with either bead capture of the mRNA or droplet barcoding (12–14). These techniques require the use of complex and expensive microfluidic systems and/or noncommercially available equipment, which is not readily accessible to most biomedical research laboratories.
Recently, we reported the engineering of a thermostable reverse transcription xenopolymerase (RTX) that enables both RT and PCR by a single enzyme (15). RTX was created by directed enzyme evolution using emulsion-based screening of large libraries within droplets containing Escherichia coli lysate. In sharp contrast to commercially available polymerases, we found that RTX is exceptionally resistant to the presence of very high concentrations of eukaryotic cell lysates. We capitalized on the cell lysate resistance of RTX to develop a very simple, high-efficiency and low-cost, one-step droplet method for NPS of immune receptors. Here, we demonstrate the utility of this technology, both for the discovery of antigen-specific antibodies and for TCRs, from human peripheral blood B cells and T follicular helper (TFH) cells respectively, following immunization with the seasonal influenza vaccine.
RESULTS AND DISCUSSION
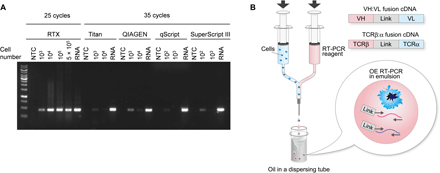
We evaluated RTX and commercially available RT-PCR kits [see also (16, 17)] side by side for their ability to produce a DNA amplicon from an RNA target in the presence of cell lysate. Fifty microliters of RT-PCRs containing detergent to mimic emulsion droplet conditions were spiked with varying quantities of human embryonic kidney (HEK) 293 cells (final cell lysate protein concentration between 4 × 10−4 and 2 μg/μl). Whereas all commercial polymerases failed to yield a visible DNA amplicon in the presence of cell lysate from 104 HEK293 cells (0.04 μg/μl) and a few generated a faint band in lysate derived from as few as 103 cells, RTX was not inhibited even in the presence of the highest concentration of lysate that could be accommodated within the 50-μl reaction volume (5 × 105 cells; Fig. 1A).

(A) Varying numbers of HEK293 cells were spiked into 50 μl of RT-PCR solution containing detergent and subjected to RT-PCR using primers to amplify the housekeeping gene PGK1. Purified HEK293 RNA was used as a positive control. NTC, no template control. (B) Two laminar flow streams, one containing a cell suspension and the other comprising the reagents required for OE RT-PCR, are merged using a Y junction to form droplets that are immediately emulsified in a dispersing tube. OE RT-PCR is performed in the emulsion to create ca 850- or 550-bp amplicons of linked VH:VL or TCRβ:α amplicons, respectively.
Previously, we developed a two-step NPS method that used an axisymmetric flow-focusing device (FFD technology) for the rapid formation of low-dispersity emulsions without microfluidics. Briefly, in this method, single cells along with oligo-dT beads and a lysis solution are first encapsulated into emulsion droplets using the FFD. After breaking the emulsion, the olido-dT beads containing captured mRNA from the lysed cells are washed and re-emulsified; then, overlap extension (OE) RT-PCR is performed to generate linked VH:VL and TCRβ:TCRα amplicons, which are sequenced by NGS to determine the paired VH:VL or TCRβ:TCRα repertoire (12).
We now developed a simple one-step emulsion system for NPS of B and T cells, which takes advantage of the exceptional lysate resistance inherent to RTX, thus circumventing the need for any specialized equipment and affording considerable technical simplicity. In short, two laminar flow streams, one containing a cell suspension and a second containing all the reagents required for OE RT-PCR (i.e., a complete primer set, RTX, buffer, and 1.4% Tween 20 for cell lysis) were mixed using a Y junction connected to a 27-gauge needle (Fig. 1B and fig. S1). The resulting droplets were immediately emulsified in a dispersion tube containing mineral oil for 5 min, giving rise to droplets with an average steady-state emulsion diameter of 3.3 ± 1.7 μm. Note that the droplet diameter is smaller than the diameter of lymphocytes because the cells are lysed during the 5-min emulsification. In-droplet OE RT-PCR was performed using 3′ primers that anneal to the constant region and either multiplex 5′ FR1 primers for VH and VL amplification or TCR V region primers (18) for TCRβ and TCRα amplification appended to linker sequences required to create a linked amplicon of the two variable domain cDNAs (fig. S2). After the emulsions were broken, the resulting VH:VL and TCRβ:α amplicons of approximately 850 and 550 base pairs (bp), respectively, were further amplified by nested PCR (representative nested PCR results in fig. S3) and sequenced using Illumina MiSeq. The raw sequence data were analyzed using a custom bioinformatics pipeline using Trimmomatic for quality filtering and MiXCR for germline gene annotation (19).
We first evaluated pairing precision of the BCR repertoire by isolating CD27+ memory B cells derived from peripheral blood mononuclear cells (PBMCs), expanding the cells for 4 days in the presence of anti-CD40 antibody, CpG oligodeoxynucleotides, interleukin-4 (IL-4), IL-10, and IL-21 (12). As an additional test, approximately 1000 B cell lymphoblast ARH-77 cells were spiked into the expanded B cell population, and then the cells were split into two aliquots, and the NPS reaction was carried out separately for each aliquot. B cell expansion produces clonal replicates for a subset of the expanded B cells, ensuring that both aliquots contain representative cells from each clonal family. When the expanded B cells are split into two aliquots and NPS is performed separately on the two aliquots, the fraction of clonal cells found in both samples [clonal cells are identified as those encoding an identical CDR-H3 (heavy-chain complementarity-determining region 3) sequence] that is paired with the same CDR-L3 (light-chain complementarity-determining region 3) can be used to calculate the pairing precision. The ratio of reads encoding ARH-77 VH linked to its cognate (natively paired) VL over those in which the ARH-77 VH was paired to an unrelated VL chain across all samples was 96.5:1 (signal-to-noise ratio; Table 1) (12). Next, the VH:VL sequences from the expanded B cells in the technical replicates observed at >1 reads were clustered at 90% CDR-H3 nucleotide sequence identity. Clustering yields high-confidence data and a conservative estimate of clonal diversity because clonally expanded or somatically mutated B cells with similar VH sequences collapse into a single CDR-H3 cluster. In donor A, the two replicates of in vitro expanded CD27+ B cells generated a similar number of VH:VL clonotypes (5761 and 5260, for the two aliquots, each containing 30,000 B cells) (Table 1, samples 1 and 1′). A total of 3166 CDR-H3 clonotypes were detected in both technical replicates, and of these, 2787 were paired with the same CDR-L3, resulting in a pairing precision of 93.8% (see Materials and Methods). The presence of B cells encoding the same antibody in both replicate aliquots is due to the partitioning of clonal cells from the same progenitor following expansion. A similar pairing efficiency and precision was observed when using expanded CD27− naïve cells from the same donor (21,801 and 17,223 VH:VL pairs in the two aliquots with pairing precision >96%). Note that a comparable ratio of clonotypes to input cells was obtained with (sample 2) or without (sample 2′) the use of SUPERase•In RNase (ribonuclease) inhibitor, the most expensive component in the reaction mixture.
Next, we compared the performance of the RTX-based NPS methodology described here with that of the well-established the FFD technology. A total of 100,000 expanded memory B cells were divided into four aliquots. Two aliquots (technical replicates) were processed using the RTX-based NPS, and two were processed using the FFD technology. We obtained 5733 and 7100 VH:VL clusters for the two technical replicates with pairing precision 89% using the RTX-NPS and 4146 and 3822 VH:VL clusters with pairing precision 87.7% using the FFD technology. The paired gene usage was essentially the same for both methods (fig. S4). The Pearson correlation coefficient of paired V gene usage comparing the two technologies was very high (averaged R value was 0.88). In summary, besides time savings and greatly simplified operation, the RTX-NPS methodology described in our paper generates very similar BCR repertoires and a slightly higher yield compared to the earlier FFD technology.
Subsequently, we determined TCR pairing precision of RTX-NPS in a similar fashion. First, 1000 Jurkat T cells were spiked into 650,000 nonexpanded PBMCs (Table 1). Following NPS and MiXCR annotation, the ratio of natively paired TCRβ:α reads to reads in which the Jurkat TCRβ was paired to an unrelated TCRα was 401:1 (signal-to-noise ratio). Second, pan T cells isolated from frozen PBMCs were incubated with anti-CD3/CD28 beads and IL-2 to expand T cells, and the expanded cell population was split into two samples and examined via NPS. The resulting TCRβ:α amplicons were sequenced, and the data were analyzed as above except that a higher CDR-β3 nucleotide sequence identity threshold was used for clustering (95% for TCRs versus 90% for BCRs, with the lower clustering threshold for BCRs necessitated by SHM). A pairing precision of ~91 to 93% was observed among technical replicates of the expanded T cells or nonexpanded PBMCs (Table 1).
We analyzed various features of the paired VH:VL and TCRβ:α repertoires determined using RTX-NPS (fig. S5). Figure S5A shows the VH and VL germline gene combinations in CD27+ and CD27− B cells. As shown in fig. S5B, the Pearson coefficients values for paired V gene usage among technical replicates were very high, highlighting the reproducibility afforded by our methodology. Note that the Pearson coefficients observed when comparing CD27+ and CD27− B cells are lower than those for technical replicates of the same B cell subset, consistent with differences in germline gene usage between memory B cells and naïve B cells reported previously (20). Similarly, TRAV-TRBV usage is shown in fig. S5C. As shown in fig. S5D, the frequency of TRBV gene detected by RTX-NPS was quite similar to that detected in the bulk TCRβ sequencing result (i.e., where only the TCRβ repertoire and not the paired repertoire is determined), suggesting that our method does not cause biased amplification for specific genes. The Pearson correlation coefficients of paired TRAV-TRBV gene usage, as well as that of paired TRAJ-TRBJ gene usage, for all possible combinations are summarized in fig. S5E. The averaged Pearson coefficients calculated among replicates from the same donors were 0.91 and 0.93 for TRAV-TRBV and TRAJ-TRBJ, respectively, highlighting the reproducibility of the data among technical replicates. The Pearson coefficients among unrelated donors showed lower values for both TRAV-TRBV and TRAJ-TRBJ gene usages, suggesting that each donor has a unique TCR repertoire, as is expected given differences in human leukocyte antigen haplotypes among individuals. As previously reported, we observed CDR3β sharing and an even greater degree of CDR3α sharing among unrelated donors (21). However, we did not observe paired CDR3β:CDR3α sequence sharing among donors (fig. S5F). Last, regarding public TCRβ:α clonotypes, as of 12 October 2019, the VDJdb database contained 20,307 human TCRβ:α clonotypes after removing duplicates. We detected only one TCRβ:α clonotype in our dataset that was also in the VDJdb.
We then evaluated the utility of our RTX-NPS methodology for the cloning of antigen specific antibodies following vaccination. Peripheral blood was collected from a healthy donor 7 days after immunization with the 2017-2018 seasonal inactivated quadrivalent influenza vaccine (Fluzone). We first sequenced the VH:VL repertoire from total B cells found in freshly isolated PBMCs. For a more representative estimation of clonal size, we separately determined the VH repertoire in CD3−CD19lo/−CD20−CD27++CD38++ plasmablasts isolated by flow cytometry (fig. S6). For influenza and for most other inactivated vaccines, the number of antigen-specific circulating plasmablasts increases markedly soon after vaccination and peaks between days 5 and 8 (22, 23). We selected the top five VH:VL sequences in PBMCs for which their VH sequences were observed at the highest number of reads in the plasmablast repertoire (thus corresponding to the most expanded B clones in the sample) for further study (table S1). The respective genes were synthesized and antibodies were expressed in Expi293F cells and purified and tested for binding to the vaccine and to historical hemagglutinin (HA) proteins (Table 2 and fig. S7). Of the five antibodies expressed, one bound to HAs from both historical and vaccine H3 strains (HT-D), one bound to HAs only from historical H3 strains (HT-B), and one bound to HAs from both historical and vaccine influenza B strains (HT-C) with subnanomolar affinities. While two other antibodies (HT-A and HT-E) did not show binding to the recombinant HA proteins, HT-E bound to the Fluzone 2017-18, suggesting that HT-E recognizes a non-HA protein such as nucleoprotein, neuraminidase, or matrix protein in the vaccine, as reported previously for a fraction of antibodies encoded by day 7 plasmablasts (22, 24, 25). Overall, the frequency of antigen-specific antibodies identified (four/five with three being HA specific) is consistent with the expected frequency of antigen-specific plasmablasts following seasonal influenza vaccination (22).
Half-maximal effective concentration (EC50) values are reported as average ± SD (n ≥ 3). H3 A/Hong Kong/4801/2014 and B/Phuket/3073/2013 are components of the 2017-2018 Fluzone vaccine.
We used an analogous approach to identify antigen-specific TCRs from peripheral TFH cells. PBMCs from blood collected at day 7 post vaccination with the Fluzone were treated with phorbol 12-myristate 13-acetate (PMA) and ionomycin and processed using RTX-NPS, yielding approximately 7000 TCRβ:α clonotypes (Table 1). A separate PBMC aliquot from the same time point was used to isolate circulating TFH cells by flow cytometry (CD3+CD4+CXCR5+CD45RA−PD1++; Fig. 2A) and subsequently, the TCRβ genes were amplified and sequenced. Two TCRβ:α sequences for which TCRβ were observed at the highest frequency in the TFH repertoire were examined further. Variable region genes were synthesized and cloned into expression vectors for producing chimeric TCRs encoding a mouse constant region and human variable region under the control of the cytomegalovirus (CMV) promoter (fig. S8), and cotransfected into Jurkat cells. Transfectants were then coincubated with IL4+GM-CSF–differentiated dendritic cells (DCs) (26) from the same donor with either antigen (i.e., Fluzone vaccine) or buffer. As a negative control, we transfected Jurkat cells with the RA14 TCR, which is specific for CMV (27). In the presence of antigen-presenting cells, Jurkat cells expressing cognate human TCRβ:α become activated resulting in increased CD69 expression, which is detected by fluorescence-activated cell sorting (FACS). One of the two TFH TCRβ:α clones tested was found to be vaccine activated and showed a marked increase in CD69 expression in samples that had been incubated with antigen (Fig. 2, B and C, and table S2).

(A) Sorting and gating strategy for follicular helper CD4+ T cells. CD3+CD4+CXCR5+CD45RA− PD1++ TFH cells were isolated from PBMCs at day 7 post vaccination from a donor vaccinated with the Fluzone vaccine. (B) Histogram showing expression of the CD69 activation marker on Jurkat cells expressing exogenous TCRs from the vaccinated donor after 24 hours incubation in the presence or absence of autologous DCs and/or Fluzone 2011-2012 vaccine. Numbers of transfected cells are shown. HT-T-1 and HT-T-2, Jurkat cells transfected with TCRβ:α from peripheral TFH cells; RA14, CMV-specific TCRβ:α as a negative control. (C) CDR3 sequences and gene usages of the HT-T-1 clone.
In summary, we discovered that RTX is extremely resistant to the presence of high concentrations of cell lysates and capitalized on this enzyme to develop a very simple droplet methodology for the NPS of both antibody and TCR repertoires. Our technique can be readily implemented without any specialized equipment to enable the sequencing of immune receptor repertoires at significant depth with a high level of precision and the discovery of antigen-specific antibodies and TCRs from human samples. We foresee that our technology may be particularly useful for the analysis of vaccine responses, including analysis of the evolution clonal lineages of neutralizing antibodies and the design of immunogens (28–30). Likewise, determination of the paired TCRβ:α repertoire is likely to be especially relevant for analyzing the repertoire in tumor-infiltrating lymphocytes for adaptive TCR therapy (13, 31). Last, we note that, in addition to NPS studies, the cell lysate–resistant RTX is likely to be a valuable tool for other single-cell applications (14, 32) as well as emulsion-based protein engineering (33).
MATERIALS AND METHODS
Recombinant expression of exonuclease-deficient RTX
The exonuclease-deficient version of RTX (N210D) was expressed and purified as described previously (15). The expression plasmid for RTX is available at Addgene.
RT-PCR with HEK293 cells using PGK1 primers
HEK293 cells were gently dissociated from the culturing plate by pipetting and washed with cold 80 mM tris-HCl (pH 7.5) twice. The cells were resuspended in cold 80 mM tris-HCl buffer (pH 7.5) within the concentration range of 500 to 1.5 × 105 cells/μl and 0.2 μl (102 to 104 cells) or 0.66 μl (105 cells) were mixed with 50 μl of various RT-PCR reagent mixtures [RTX; Titan One Tube RT-PCR System (no. 11855476001; Sigma-Aldrich), QIAGEN OneStep RT-PCR Kit (no. 210210; QIAGEN), SuperScript III One-Step RT-PCR System (no. 12574-026; Thermo Fisher Scientific), and qScript One-Step qRT-PCR Kit (no. 95039-940; VWR)] containing 0.5% Tween 20. To perform the experiment with 5 × 105 cells, the buffer of cell suspension was replaced with cold 1× RTX buffer and 3.3 μl of 1.5 × 105 cells/μl cell suspension was mixed with 50-μl RTX reagent mixtures. Detailed RT-PCR reagent recipes and protocols are described in table S3. Total HEK293 cell RNA (300 ng) was used as a positive control. Phosphoglycerate kinase 1 (PGK1) primers (forward, AGGTGCTCAACAACATGGAGA; and reverse, CCCCAGTGCTCACATGGCTGACTTT) were designed to produce a 450-bp amplicon. All primers in this study were purchased from Integrated DNA Technologies or Sigma-Aldrich and dissolved in ultrapure water.
Setup for RTX-based NPS
To construct the Y junction, Tygon tubes (no. 80-10002-03, 1.5875 mm inner-diameter; Cytek Biosciences) were connected to syringes via female Luer Lock to barb connectors (no. 11532; Qosina) on one end and a barbed Y connector on the other (polyvinylidene difluoride, 1.5875 mm inner-diameter; no. 3063342, Cole-Parmer). A short silicone tube (no. 8060-0020, 1.5875 mm inner-diameter; Thermo Fisher Scientific) was used to connect the Y connector outlet to a 27-gauge needle (fig. S1).
NPS analysis
To analyze the T cell receptor repertoire, cells were stimulated or expanded before NPS as described below. Cells were centrifuged at 300g for 10 min at 4°C, and the supernatant was removed completely. The cells were resuspended in cold 300-μl 2× RTX buffer [120 mM tris-HCl (pH 8.4), 50 mM (NH4)2SO4, 20 mM KCl, and 2 mM MgSO4] and centrifuged at 900g for 5 min at 4°C. The supernatant was completely removed, and cold 1-ml 2× RTX buffer was added without resuspension and then centrifuged at 900g for 3 min at 4°C. The supernatant was removed, and the cells were resuspended in cold 2× RTX buffer at the concentration range from 7.9 × 104 to 2 × 105 cells/ml and loaded into a syringe. RTX reagent described in tables S4 to S6 was loaded into a second syringe and chilled on ice. Emulsification oils were prepared as follows, and either Abil EM 90–based oil or Span 80–based oil was used for the experiments: Abil EM 90–based oil [mineral oil (no. M5904; Sigma-Aldrich) containing 2% Abil EM 90 (Degussa), and 0.05% Triton X-100 (no. T8787; Sigma Aldrich)] and Span 80–based oil [mineral oil containing 4.5% Span 80 (no. S6760; Sigma-Aldrich), 0.4% Tween 80 (no. P8074; Sigma-Aldrich), 0.05% Triton X-100, v/v%)]. Oils were mixed by gentle inversion, and 11.3 or 31 ml was measured into a DT-20 or DT-50 tube (nos. 0003703100 and 0003699600; IKA) and chilled on ice. The syringes were connected to a Y junction, and a syringe pump (KD Scientific Legato 200, Holliston, MA, USA) was used to simultaneously expel both the cells and reagents at 1.3 ml/min into the cold oil dispersed with DT-20 or DT-50 tube on ULTRA-TURRAX Tube Drive (IKA) at 615 rpm. The final aqueous to oil ratio was 1:3.1. Dispersion continued for 5 min after the stream injection had been completed. The emulsion was aliquoted into 96-well plates at 100 μl per well, and OE RT-PCR was performed under the following conditions: 30 min at 68°C, 2 min at 94°C, followed by 25 cycles of PCR amplification (94°C for 30 s, 60°C for 30 s, and 68°C for 2 min) and cDNA extension at 68°C for 7 min.
Following the RT-PCR, emulsions were collected in 2-ml microcentrifuge tubes and centrifuged at 17,000g for 10 min at 4°C. The supernatant was decanted, and the DNA was extracted as described previously (19). Briefly, the Span 80–based oil required two extractions with water-saturated diethyl ether, whereas the Abil EM 90–based oil required serial extractions using water-saturated diethyl ether, water-saturated ethyl acetate extraction, and water-saturated diethyl ether in order. Ether was evaporated, the sample was diluted fivefold into DNA binding buffer, column concentrated (no. C1003-50, no. D4004-1-L, and no. D4003-2-48; Zymo Research), and eluted into 50 μl of ultrapure water. In volumes greater than 3 ml, the aqueous phase was EtOH-precipitated and then column-concentrated. During NPS of TCRs, cDNA was purified using AMPure XP beads (no. A63880; Beckman Coulter) according to the manufacturer’s instructions at a 2:1 (cDNA:beads) ratio and reconstituted in 50 μl of ultrapure water.
Nested PCR was performed on 20 to 30% (BCR) or 5 to 10% (TCR) of the cDNA in a total volume of 250 μl with DreamTaq Hot Start DNA Polymerase (no. EP1702; Thermo Fisher Scientific), using primers described in tables S7 (BCR) and S8 (TCR) under the following conditions: 94°C for 3 min, followed by 25 to 30 cycles of PCR amplification (94°C for 30 s, 62°C for 30 s, and 72°C for 1 min) and a final extension of 72°C for 7 min. The ~850-bp VH:VL amplicon and ~550-bp TCRβ:α amplicon were then gel-purified (no. C1003-50, no. D4001-1-100, and no. D4003-2-48; Zymo Research).
A two-step procedure was performed to append Illumina adaptor sequences to the BCR amplicon. First, 50 ng of DNA was amplified using NEBNext High-Fidelity 2× PCR Master Mix (New England Biolabs Inc.) in combination with the primers in table S9 and the conditions described in table S10. The PCR product was column-concentrated (Zymo Research) and quantified by NanoDrop. In the second reaction, 50 ng of DNA was amplified in combination with the primers in table S11 under the same conditions using eight cycles for the amplification. The ~1000-bp PCR product was gel-purified (Zymo Research). A one-step procedure was performed to append Illumina adaptor sequences to the TCR amplicon. Fifty nanograms of DNA was amplified with the primers in table S11 under the same conditions using six cycles for the amplification. The ~650-bp PCR product was gel-purified. The DNAs were submitted for Illumina MiSeq 2×300 sequencing. Once the DNA products are barcoded with primers described in table S11, the barcoded DNAs are pooled for sequencing to reduce the cost. We typically apply 500,000 reads for each sample. It is recommended that additional sample-specific barcode sequences are added to the nested primers (tables S7 and S8) when the MiSeq chip is full of BCR/TCR amplicons. In our laboratory, we routinely achieve a total NPS running cost of <$280 per ~104 paired clonotypes including sequencing cost.
Bioinformatic analysis
The BCR/TCR repertoire was analyzed as described previously (19). Briefly, sequences were quality filtered using Trimmomatic by trimming sequences following a 5-bp stretch with an average Phred score <20 and annotated by MiXCR software (34, 35). VH:VL pairs, TCRβ:α pairs with greater than one read were clustered at 90% CDR-H3 nucleotide identity and 95% CDR-β3 nucleotide identity, respectively. We assigned highest-frequency CDR-L3, CDR-α3 in each cluster as the correct partner for CDR-H3, CDR-β3, respectively. The pairing precision was calculated with the following formula as described before (12, 19) and the results are shown in Table 1.P=TP1 and 2TP1 and 2+FP1 or 2
TP1 and 2 is the number of CDR-H3 amino acid sequences paired with identical CDR-L3 amino acid sequences in both replicates. FP1 or 2 is the number of CDR-H3 amino acid sequences paired with different CDR-L3 amino acid sequences across the replicates. P is the VH:VL pairing precision. In case of TCR pairing precision, CDR-H3 was replaced with CDR-β3, and CDR-L3 was replaced with CDR-α3.
Cell culture
All media except Expi293 Expression Media in this study contained 1× penicillin/streptomycin (Life Technologies). HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS). Expi293F cells were cultured in Expi293 Expression Medium, and all lymphocytes, ARH-77 cells, and Jurkat cells were cultured in RPMI 1640 medium containing 10% FBS, 2 mM l-glutamine, 1× nonessential amino acids, and 1× sodium pyruvate at 37°C in the presence of 5% CO2. Several additional reagents were used to stimulate or expand cells as described below.
Cell isolation, expansion, and stimulation
This study was approved by the University of Texas at Austin Institutional Review Board (2012-08-0031) and Institutional Biosafety Committee (2016-00045). Unless otherwise indicated, PBMCs were isolated using Lymphocyte Separation Medium (density, 1.077 g/liter; no. 12002-030; VWR) from donated human whole blood after informed consent had been obtained (Gulf Coast Regional Blood Center, Houston, TX). The PBMCs were cryopreserved using RPMI 1640 medium containing 10% FBS and 10% dimethyl sulfoxide and stored in liquid nitrogen. To expand B cells, frozen PBMCs were thawed, and CD27+ memory B cells were isolated with a Memory B Cell Isolation kit (no. 130-093-546; Miltenyi Biotec). CD27− B cells, which passed through an MS Column (no. 130-042-201; Miltenyi Biotec), were also collected. B cells were expanded for 4 days in the presence of anti-CD40 antibody (10 μg/ml; 5C3, BioLegend), CpG ODN 2006 (1 μg/ml; InvivoGen), IL-4 (100 U/ml), IL-10 (100 U/ml), and IL-21 (50 ng/ml; PeproTech). To expand T cells, frozen PBMCs were thawed, and total T cells were isolated with a Pan T cell isolation kit (no. 130-096-535; Miltenyi Biotec) and expanded with CD3/CD28 dynabeads (no. 11161D; Thermo Fisher Scientific) and IL-2 (30 U/ml; PeproTech) for a week. The medium was exchanged every 3 days, and fresh beads and IL-2 were added. For the analysis of influenza vaccine response, PBMCs were isolated from a healthy 25-year-old human donor (donor C) 7 days after vaccination with Fluzone Quadrivalent vaccine, with informed consent. One million PBMCs were directly used for NPS of the VH:VL. For the TCR repertoire analysis, 650,000 PBMCs were stimulated with PMA (100 ng/ml; no. P8139s, Sigma-Aldrich) and ionomycin (100 ng/ml; no. I9657; Sigma-Aldrich) for 4 hours and then subjected to NPS directly. Technical replicates with a spike-in control of 1000 Jurkat cells were performed without SUPERase•In RNase inhibitor (Table 1, samples 5′). FACS was used to isolate plasmablasts and memory B cells. The remaining PBMCs were cryopreserved and used for TFH cells isolation.
FACS of plasmablasts, memory B cells, and TFH cells
PBMCs were stained with anti-human CD19-v450 (HIB19; BD Biosciences, San Jose, CA), CD27-APC (M-T271; BD Biosciences), CD38-PE (HIT2; BioLegend, San Diego, CA), CD20–fluorescein isothiocyanate (FITC) (2H7; BioLegend), and CD3-PerCP/Cy5.5 (HIT3a; BioLegend). CD3−CD19+CD20+CD27+ memory B cells and CD3−CD19lo/−CD20−CD27++CD38++ plasmablasts were sorted directly into 1 ml of TRIzol reagent (Thermo Fisher Scientific) using a FACSAria Fusion cell sorter and BD FACSDiva 8.0.1 software (BD Biosciences) (fig. S6). For TFH cell isolation, frozen PBMCs were thawed and stained with anti–PD1-PE (no. 12-2799-41; Thermo Fisher Scientific), anti-CXCR5-BB515, anti-CD3− Alexa Fluor 700, anti–CD4-Pacific Blue, and anti-CD45RA− PE-CF594 (no. BDB564625, no. BDB561027, no. BDB558116, and no. 562326; BD Biosciences, respectively) antibodies, and then CD3+CD4+CXCR5+CD45RA− PD1++ TFH cells were sorted directly into 1 ml of TRIzol reagent (Fig. 2A).
RNA isolation and cDNA synthesis for VH-only and TCRβ-only sequencing
We purified RNA from the TRIzol-lyzed cells by using RNeasy Mini Kit (no. 74104; QIAGEN). RNA from TFH (250 ng), plasmablasts (500 ng), and memory B cells (500 ng) was reverse-transcribed and amplified as described previously (36). TCRβ cDNA was amplified using KAPA HiFi HotStart Real-time PCR Master Mix (no. KK270; Kapa Biosystems), TRBV primers described in table S6, and the TRBC primer ACCAGTGTGGCCTTTTGGGTGTG. The PCR was carried out under the following conditions: 98°C for 45 s; 98°C for 15 s, 60°C for 30 s, 72°C for 1 min × 35 cycles; and 72°C for 7 min. The resulting TCRβ DNA was column-purified (Zymo Research) and amplified for eight cycles using primers described in table S12 (18). The VH and TCRβ DNAs were gel-purified (Zymo Research) and submitted to the Genome Sequencing and Analysis Facility at The University of Texas at Austin and sequenced with Illumina MiSeq 2×300.
Enzyme-linked immunosorbent assay
Selected VH:VL pairs from plasmablasts/memory B cells (table S1) were cloned, expressed, purified, and characterized using enzyme-linked immunosorbent assay as described before (25). The following recombinant HA proteins and a vaccine were obtained through BEI Resources, National Institute of Allergy and Infectious Diseases, National Institutes of Health: HA A/Wisconsin/67/2005 (H3N2), NR-49237; HA A/New York/55/2004 (H3N2), NR-19241; HA B/Florida/4/2006, NR-15169; HA B/Brisbane/60/2008, NR-19239; and Fluzone Quadrivalent Influenza Vaccine, 2017-2018, NR-51401. HA A/Michigan/45/2015 (H1N1) was purchased at eENZYME (no. IA-H1-M15WP). HA A/Hong Kong/4801/2014 X-263B (H3N2) and HA B/Phuket/3073/2013 were gifts from S. C. Harrison.
Construction and transfection of TCR plasmids
TCRβ sequences isolated from TFH were matched to the TCRβ:α repertoire using CDR-β3 sequence homology. Selected TCRβ and TCRα were cloned into modified pcDNA3.1 expression vectors harboring mouse TCR constant regions and mouse TCR leader peptide sequences (fig. S8 and table S2). A total of 7.5 μg of TCRβ and 7.5 μg of TCRα plasmids were electroporated into 2 × 107 Jurkat cells using Gene Pulser Xcell Electroporation Systems using 250 V and 950 μF, which have a ~27.5-ms time constant. After 24 hours of incubation, Jurkat cells were used in the antigen specificity assay.
Analysis of TCR antigen specificity
PBMCs were isolated eight months after the Fluzone vaccination. DCs were prepared from the PBMCs as described before (26). DCs were dissociated by using trypsin-EDTA and washed. Transfected Jurkat cells were washed, and then 4 × 104 Jurkat cells were mixed with or without 4 × 104 DCs in a total volume of 200-μl RPMI 1640 medium containing 5% human serum type AB (no. H4522; Sigma-Aldrich) and with or without 2.5-μl Fluzone Vaccine 2011-2012 (no. NR-36747; BEI resources) and incubated for 24 hours in a 96-well plate. The cells were stained at 4°C for 20 min in phosphate-buffered saline/0.2% bovine serum albumin/2 mM EDTA with FITC anti-human CD69 (no. 310904; BioLegend), PE anti-mouse TCRβ chain (no. 109207; BioLegend), and PerCP-Cy 5.5 anti-human CD3 (no. 552852; BD Biosciences). Cells were washed and analyzed with BD FACSAria and FlowJo v10 software.
SUPPLEMENTARY MATERIALS
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial license, which permits use, distribution, and reproduction in any medium, so long as the resultant use is not for commercial advantage and provided the original work is properly cited.
REFERENCES AND NOTES
Acknowledgments: We thank R. Salinas for assistance with FACS; S.C. Harrison (Harvard Medical School) for HA proteins; and the Genome Sequencing and Analysis Facility at the University of Texas at Austin for performing Illumina sequencing. Funding: Funding for this work was provided by U.S. Defense Threat Reduction Agency (DTRA) HDTRA1-12-C-0105 (G.G.) and HDTRA1-18-1-0030 (A.D.E.); National Institutes of Health (NIH) grant 5R21CA191239-02 (A.D.E.) and grant U19 AI057266 (G.G. and J.J.G.); Texas Health Catalyst program no. 2056003826 (G.C.I.); and the Welch Foundation (F-1654 to A.D.E. and F-1767 to J.A.M.). H.T. was supported by UTHealth Innovation for Cancer Prevention Research Training Program Post-Doctoral Fellowship (Cancer Prevention and Research Institute of Texas grant no. RP160015), Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowships for Research Abroad, and Uehara Memorial Foundation Research Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Cancer Prevention and Research Institute of Texas. Author contributions: H.T., J.R.M., G.C.I., A.D.E., and G.G. developed the methodology. H.T., J.R.M., G.C.I., A.D.E., and G.G. wrote the manuscript. H.T., J.R.M., C.A.S., G.C.I., and G.G. designed the experiments. H.T., J.R.M., C.A.S., W.N.V., J. Li, J.Le., J.J.G., Y.T., G.D., A.P., and J.W.E. performed the experiments. H.T., J.R.M., and R.D. conducted bioinformatic analyses. All authors edited and approved the manuscript. Competing interests: H.T., J.R.M., G.C.I., A.D.E., and G.G. are inventors on a patent application related to this work, which has been submitted by the University of Texas at Austin (international patent application no. PCT/US2018/044171 based on U.S. serial no. 62/537,686). All other authors declare that they have no competing interests. Data and materials availability: NGS datasets have been deposited in the NCBI Sequencing Read Archive (SRA) under accession number PRJNA596245. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.